






















先天性直肠肛门畸形

什么是先天性直肠肛门畸形
先天性直肠肛门畸形(anorectum development malformation inborn)多见,并且类型众多,直肠盲端和瘘管的位置各异。其发病率在新生儿中为1∶1500~5000,占消化道畸形的首位。男性多于女性,高位畸形在男性约占50%,女性占20%。各种瘘管的发生率在女性为90%,男性为70%。合并其它先天性畸形的发生率约有30~50%,且常为多发性畸形。有家族史者少见,仅1%。有遗传性,但遗传方式尚无定论。
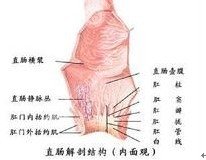
先天性直肠肛门畸形的临床症状
婴儿出生后仔细观察会阴部即可发现在正常肛门位置没有肛门,特别是生后24小时不排胎便,就应及时检查。如能早期发现,其临床表现为不同程度的低位肠梗阻症状。在无瘘的或伴有狭小瘘管的病例,往往在出生后早期即发生急性性低位肠梗阻症状。在肛门直肠狭窄或伴有较大瘘管的病例,依据其狭窄程度和瘘管大小,可在几周、几月甚至几年后出现排便困难、便秘、粪石形成、继发巨结肠等慢性梗阻症状。
(1)高位畸形:约占40%,男孩多见,往往有瘘管存在,但因瘘管细小,几乎都有肠梗阻症状。骨盆肌肉的神经支配常有缺陷,并伴有骶椎和上尿路畸形。此型病例在正常肛门位置皮肤稍凹陷,色素较深,但无肛门。哭吵时凹陷处不向外膨出,用手指触摸也无冲击感。女孩往往伴有阴道瘘,开口于阴道后壁弯窿部。外生殖器亦发育不良,呈幼稚型,粪便经常从瘘口流出,易引起生殖道感染。男孩常伴有泌尿系瘘,从尿道口排出气体和胎便,可反反复生尿道炎、阴茎头炎和上尿路感染。
(2)中间位畸形:约占15%。无瘘者直肠盲端位于尿道球部海绵体肌旁或阴道下段附近,耻骨直肠肌包绕直肠远端。有瘘者其瘘管开口于尿道球部、阴道下段或前庭部。其肛门部位的外观与高位畸形相似,也可以从尿道或阴道排便。探针可以通过瘘口进入直肠,在会阴部可触到探针的顶端。在女孩以直肠前庭瘘多见,因瘘口位于阴道前庭舟状窝部,也称舟状窝瘘,瘘孔较大,婴儿早期通过瘘孔基本能维持正常排便,可引起阴道炎或上行性感染。
(3)低位畸形:约占40%。直肠末端位置较低,多合并有瘘管,很少伴发其它畸形。有的在正常肛门位置为薄膜所覆盖,隐约可见胎便色泽,哭吵时隔膜明显向外膨出,有时肛膜已破,但不而排便困难。在男孩伴有肛门皮肤瘘,管中充满胎便而呈深蓝色,瘘口位于会阴部,或更前至阴囊缝,或尿道尾侧的任何部位。在女孩伴有肛门前庭瘘或皮肤瘘,瘘口位于阴道前庭部或会阴部。
先天性直肠肛门畸形的治疗
外科治疗的目的是重建具有正常控制功能的排便肛门,方法和时间的选择,根据各种不同的类型和合并瘘管的情况而定。其治疗原则是为了改善术后排便控制功能,拖出的直肠必须通过耻骨直肠肌环,为了更好地识别耻骨直肠肌和尿道,中间位和高位畸形可采用经骶尾路肛门成形术或经骶腹会阴肛门成形术。手术时尽可能减少盆腔神经的损伤以增进感觉,拖下的直肠必须血供良好,无张力地到达会阴,缝合时使皮肤卷入肛内以防止粘膜脱垂等,这些都是要点,手术者有无此种概念,将决定预后是否良好。现今多数医师主张不适合会阴肛门成形术者,生后均先行暂时性结肠造瘘术,待至6~10个月时施行肛门成形术,术后3个月关闭造瘘。
后矢状切口的手术方法,分离到直肠盲端,在直视下处理瘘管,以电刺激识别肌群的位置,保存直肠及肛周的肌肉神经血管组织,并恢复原状,如若直肠太短或太宽,则从腹腔游离及作尾状修剪,使直肠盲端通过肛提肌及括约肌群中央,从而得到满意的排便功能。
低位畸形的治疗,如肛门皮肤瘘无狭窄,排便功能无障碍者,不需治疗。肛门或直肠下端轻度狭窄,一般采用扩张术多能恢复正常功能。对肛门皮肤瘘者,仅作简单的"后切"手术。膜性肛门在新生儿期施行会阴肛门成形术。肛门前庭瘘如瘘孔较大,在一段时间内尚能维持正常排便,可于6个月以后施行手术。低位者因已通过耻骨直肠肌环,故手术较为容易,且术后排便功能良好。
泄殖腔畸形的治疗,因一穴肛畸形复杂、新生儿期先作暂时性结肠造瘘,6月~1岁时行术。作成皮管或带蒂小肠移植的阴道成形术,在新阴道后方行腹会阴肛门成形术,利用原泄殖腔构成尿道一部分,进行泌尿系器官成形术,争取一期完成。
福州医博肛肠医院有限公司责任编辑:Dongdong48.7%患者选择了:咨询肛肠医生
![]() 在线医生坐诊,快速诊断病情
在线医生坐诊,快速诊断病情
44.5%患者选择了:直接挂医生号
![]() 直接网上挂号,就医无需排队
直接网上挂号,就医无需排队
为贯彻两会"医改"方针,方便患者寻医解疑尽早病愈,我院现已开展网上预约就诊绿色通道:
★ 网上在线咨询 ★
有医博肛肠医师在线一对一解答患者问题,并根据您的病情给予专业的个性化指导意见,提供专业治疗方案,严格保密。★ 网上预约挂号 ★
网上预约免医生挂号费,免排队等待时候,到院直接就诊。★ 假日医师门诊 ★
方便患者节假日看病,不影响上班,提供人性化服务!





















 闽公网安备 35010402350225号
闽公网安备 35010402350225号